This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
What is phylogenetics?
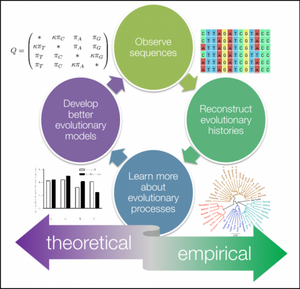
Phylogenetics is the study of evolutionary relationships among species, individuals, or genes. [1] Phylogenetic trees can be made to explore evolutionary histories and learn more about evolutionary processes. The more we learn from these trees and histories, the more we can develop even better models to explore phylogeny further. The figure to the right lays out an overview of this process.
Several methods exist for constructing phylogenetic trees inclulding Maximum Likelihood, Neighbor-Joining, and Average Distance. These methods are described further below.
Several methods exist for constructing phylogenetic trees inclulding Maximum Likelihood, Neighbor-Joining, and Average Distance. These methods are described further below.
Methods for constructing phylogenetic trees
Maximum Likelihood
In this method, an initial tree is first built using a fast but suboptimal method such as Neighbor-Joining From there, its branch lengths are adjusted to maximize the likelihood of the data set for that tree topology under the desired model of evolution. This method is advantageous because all of it's models are explicit, so they can be evaluated and improved. [2]
Neighbor-Joining
This method essentially uses the similarity scores generated by percent identity or BLOSSUM to determine which species are most closely related. It then calculates the branch lengths and draws a tree. [2]
Average Distance
This method uses the similarity scores to determine which species are most closely related and joins them with equal branch lengths, assuming that both species have diverged equally from the common ancestor. [2]
In this method, an initial tree is first built using a fast but suboptimal method such as Neighbor-Joining From there, its branch lengths are adjusted to maximize the likelihood of the data set for that tree topology under the desired model of evolution. This method is advantageous because all of it's models are explicit, so they can be evaluated and improved. [2]
Neighbor-Joining
This method essentially uses the similarity scores generated by percent identity or BLOSSUM to determine which species are most closely related. It then calculates the branch lengths and draws a tree. [2]
Average Distance
This method uses the similarity scores to determine which species are most closely related and joins them with equal branch lengths, assuming that both species have diverged equally from the common ancestor. [2]
StAR Phylogenetic Tree Construction
1. Compile protein sequences in FASTA format for each homolog
2. Align sequences using MEGA
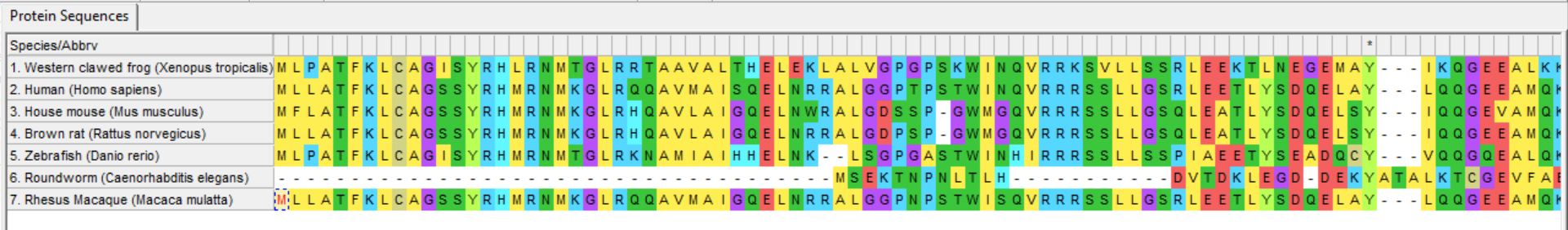
3. Build trees using MEGA
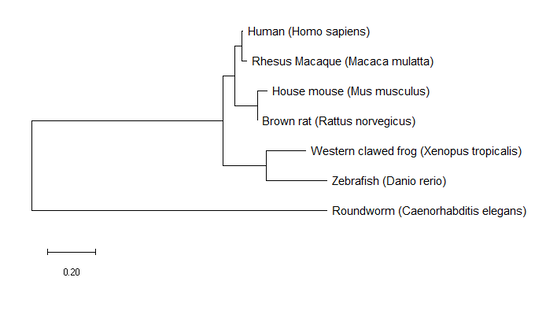
Maximum Likelihood
|
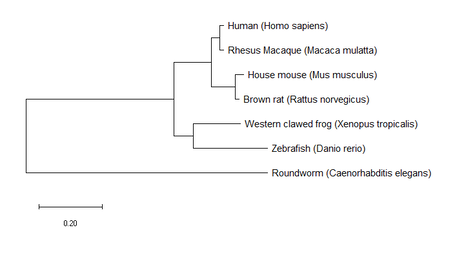
Neighbor-Joining
|
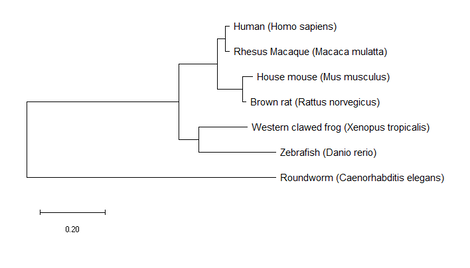
Minimum Evolution
Conclusion
All three trees constructed by MEGA for StAR homologs are very similar. The only slight difference is some of the branch lengths, but the nodes and following branches are all identical. These results agree with the listed identities for each homolog in relation to humans on my Homology page in that rhesus macaque had the highest identity value at 97% and Roundworm had the lowest at 28%. These trees provide us with information on the evolutionary relationships of StAR between the included homologs.
References
[1] What is phylogenetics? https://www.ebi.ac.uk/training/online/course/introduction-phylogenetics/what-phylogenetics
[2] Yang, Z. & Rannala, B. (2012). Molecular phylogenetics: principles and practice. Nature Reviews Genetics. 13:303-314.
[1] What is phylogenetics? https://www.ebi.ac.uk/training/online/course/introduction-phylogenetics/what-phylogenetics
[2] Yang, Z. & Rannala, B. (2012). Molecular phylogenetics: principles and practice. Nature Reviews Genetics. 13:303-314.
Header image: http://wpnature.com/small-hill-lovely-forest-canopy-leaves-unnamed-colorful-wallpapers/
